The extract command processes genetic barcoding data from raw sequencing files or BAM files generated by a wide array of technologies.
It requires two input files:
- YAML Configuration File: Specifies the parameters for the extraction process.
- CSV Inputs File: Defines the file paths for the sequencing data.
Outputs¶
- Count matrices for each sample, saved separately.
- A final merged count matrix combining all samples in anndata and csv formats.
Usage¶
On a terminal run the command as follows:
Input Files¶
YAML File Example¶
This file specifies all the running parameters for the extract pipeline. Depending on the parameters provided, QuiCAT will either use the reference-free or reference-based workflow, enable sequencing error correction, and manage error tolerance during the extraction or alignment phases of the respective workflows.
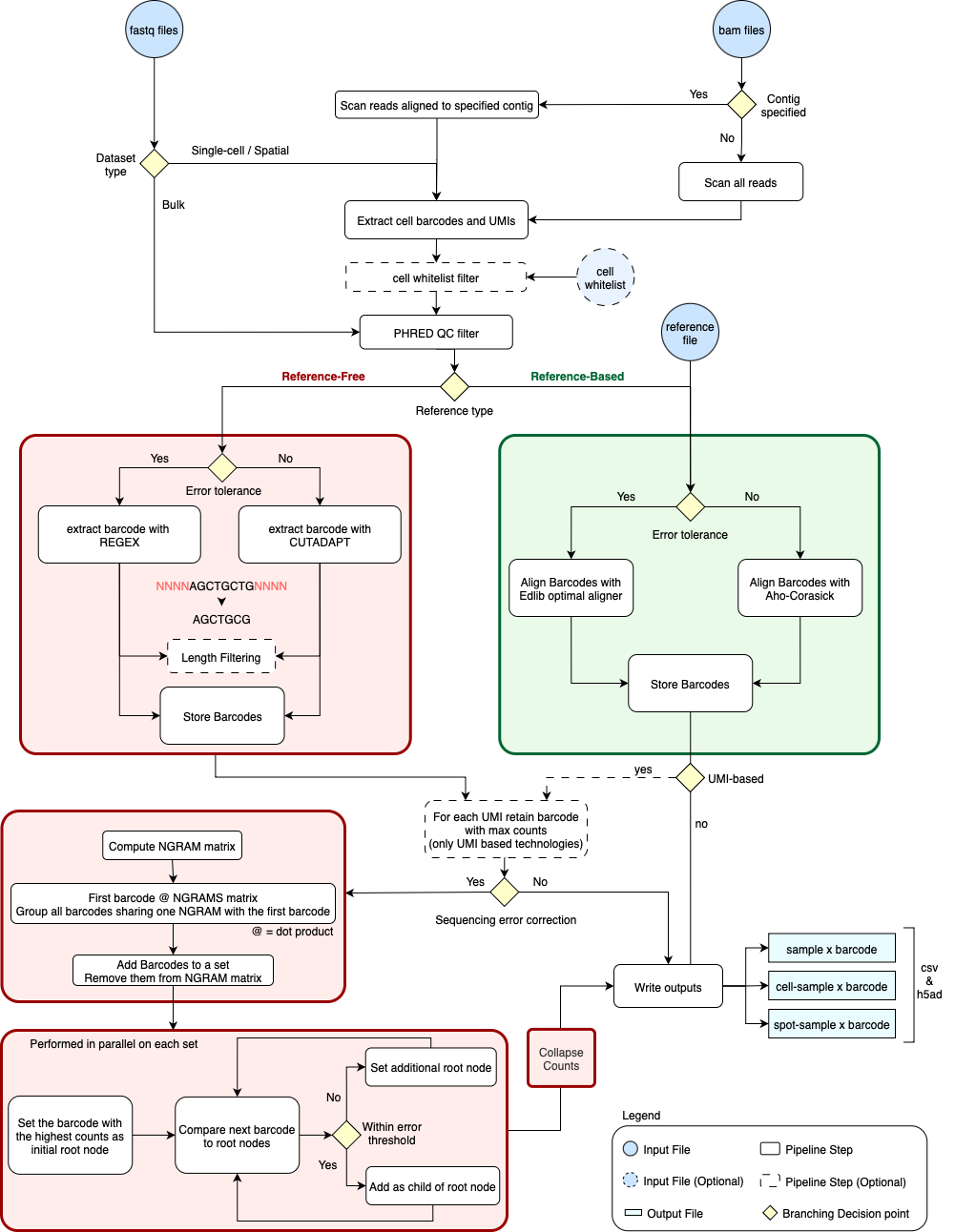
You can find a high-level overview of the QuiCAT extract workflow and how the specified parameters guide the software’s decision-making during execution in the figure below.
config:
sequencing_technology: (str) "10x"
input: (str) "bam"
paired_end: (bool) false
barcodes_path: (bool) false
phred33: (bool) true
read_qc_threshold: (int) 20
read_qc_percentage: (int) 80
filter_barcodes_relative_abundance: (float) 0.001
filter_barcodes_raw_numbers: (int) null
reference: (str) "GCTACTTGAT*ATCCTACTTG"
contig: (str) '*'
flanked_pattern: (bool) true
min_read_length: (int) 40
max_read_length: (int) 40
read_length: (int) 40
aln_mismatches: (int) null
flanking_mismatches: (float) null
left_flanking_coverage: (int) null
right_flanking_coverage: (int) null
distance_threshold: (int) 8
barcode_ratio: (int) 5
n_threads: (int) -1
input_csv: (str) "/path/to/input.csv"
output_path: (str) "/path/to/output"
YAML Parameters: in-depth explanation¶
config section¶
general parameters¶
| Parameter | Description | Default Value |
|---|---|---|
sequencing_technology |
Technology (e.g., "10x", "Parse", "DNA"). |
"10x" |
input |
File format ("fastq" or "bam"). |
"bam" |
paired_end |
Set true for paired-end reads (only for fastq). |
false |
threads |
Set the threads to use, default to using all available threads. | -1 |
QC thresholds¶
| Parameter | Description | Default Value |
|---|---|---|
read_qc_threshold |
Minimum quality score. | 20 |
read_qc_percentage |
Minimum percentage of bases that meet the quality score. | 80 |
phred33 |
Set true for Phred+33 or false for Phred+64. |
true |
barcodes filters¶
| Parameter | Description | Default Value |
|---|---|---|
filter_barcodes_relative_abundance |
In the final count matrix only retain barcodes with relative abundance above this value. | 0.001 |
filter_barcodes_raw_numbers |
In the final count matrix only retain barcodes with raw count above this value. | null |
Note
Frequencies or raw counts intended over individual samples.
barcodes extraction¶
| Parameter | Description | Default Value |
|---|---|---|
reference |
Sequence for alignment (string or path to a file). | |
contig |
Contig to extract from BAM files ('*' for unmapped reads). |
'*' |
flanked_pattern |
true if the reference specify a flanked_pattern e.g. seq1*seq2 |
false |
aln_mismatches |
If specified in conjuction with a reference file, switch from aho-corasick to optimal aligner | 0 |
flanking_mismatches |
If specified in conjuction with a flanked reference, switch from regex to cutadapt | 0.0 |
left_flanking_coverage |
Minimum base pairs overlap on the left flanking sequence | 0 |
right_flanking_coverage |
Minimum base pairs overlap on the right flanking sequence | 0 |
flanked_pattern |
true if the reference specify a flanked_pattern e.g. seq1*seq2 |
false |
min_read_length |
Minimum barcode length to retain. | null |
max_read_length |
Maximum barcode length to retain. | null |
read_length |
Fixed barcode length to retain. | null |
Note
Use min_read_length and max_read_length in combination to specify a range.
barcodes collapsing¶
| Parameter | Description | Default Value |
|---|---|---|
distance_threshold |
Distance theshold for barcode collapse. If set to 0, barcodes are not collapsed | null |
barcodes_ratio |
Minimum fold for a barcode to be collapsed into one with more counts. | null |
I/O¶
| Parameter | Description | Default Value |
|---|---|---|
input_csv |
Path to csv file with input files paths. See below. | null |
output_path |
Path to the folder where to store the pipeline's output. | null |
CSV File Example¶
Specify the input files path and metadata through the input csv file. The file must have the following columns:
Explanation:
| Parameter | Description | Notes |
|---|---|---|
sample |
Sample name as added in the final count matrix. | |
fastq_path_r1 |
Path to R1 fastq file, or only fastq file if not paired end. | Leave blank if using bam |
fastq_path_r2 |
Path to R2 fastq file. | Leave blank if not paired end |
barcodes_path |
Path to barcodes.tsv.gz if doing (single cell or spatial only). |
Leave blank if no cell/spot whitelist is needed |
bam_path |
Path to bam file. | Leave blank if using fastq |
condition |
Specify disease or treatment status (added to final count matrix) | Optional |
replicate |
If the sample is a technical replicate, specify the general sample name here (added to final count matrix) | Optional |