Extract Tutorial¶
Outline¶
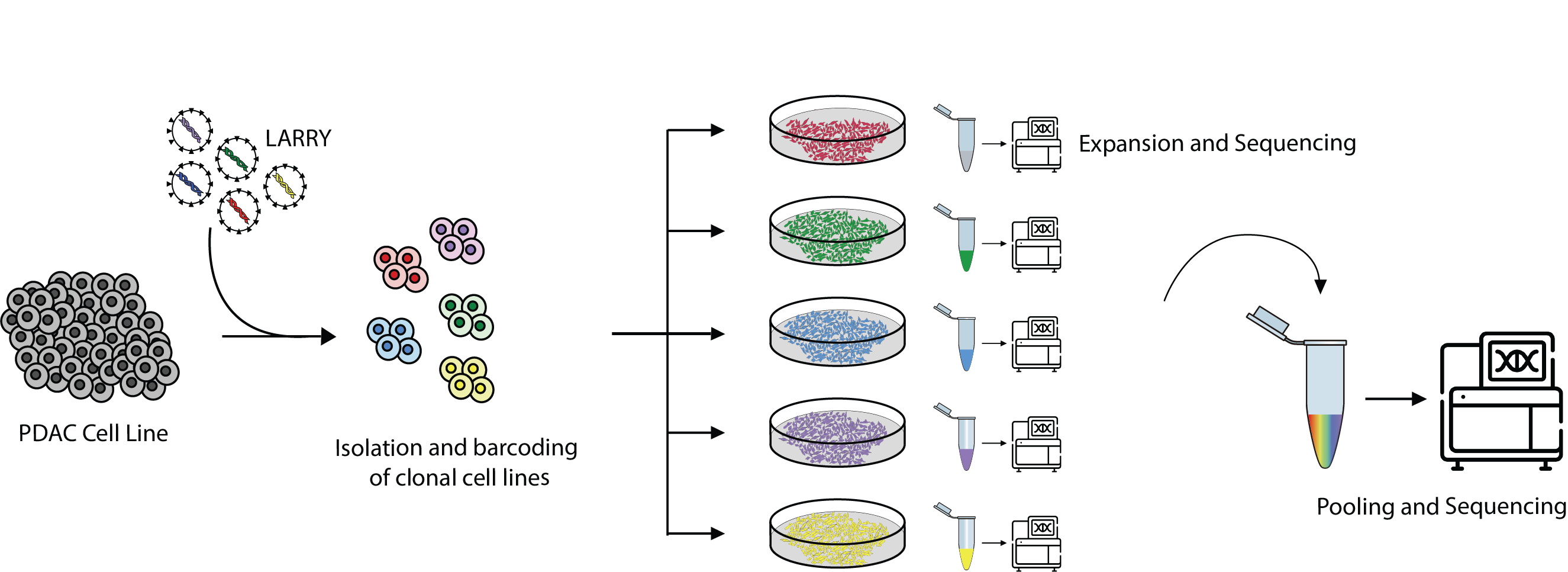
This notebook shows how to create the necessary configuration files to run QuiCAT extract workflows. For demonstration purposes, we will use a sample with bulk DNA sequencing of cell lines that were separately barcoded using a modified version of LARRY, and then pooled together. Each cell line was tagged with a unique barcode and sequenced individually before pooling to confirm the sequences (Figure 1).
In this scenario, the best approach would be to use the reference-based method of QuiCAT, but we will showcase both reference-free and reference-based approaches for demonstration purposes.
The modified version of the LARRY barcode has the following structure:
NNNN CT NNNN AC NNNN followed by a conserved region CATA
We will show how to extract this barcode using:
- A reference-free approach exploiting the conserved flanking region
- A reference-free approach leveraging the fixed nucleotide positions in the barcodes
- A reference-based approach
We will explain the input fields of the YAML configuration file in detail, populating it step by step as we go.
Finally, for the reference-based approach, we will provide the reference FASTA file at the end of this tutorial. However, you can also apply what you’ve learned to restart from the single-clone sequencing files and generate the reference yourself.
Dataset Download¶
The dataset used in this tutorial can be downloaded from Zenodo:
cd desired_download_location
wget -O larry_dna.tar.gz https://zenodo.org/records/16911389/files/larry_dna.tar.gz?download=1
tar -xzvf larry_dna.tar.gz
cd larry_zenodo
After the download and extraction of the archive is done you will find 2 folders:
single_clones: contains the FASTQ files of the individually sequenced clonespooled_clones: contains the FASTQ files from the pooled clone sequencing experiment
Note: single_clones data is single-end sequencing, while the pooled_clones data is paired-end sequencing. Keep this in mind if you want to restart from the single-clone stage to generate the reference.
Configuration file overview¶
The QuiCAT extract CLI command requires a yaml configuration file to specify the parameterers for the extraction process.
This section explains how to compose the YAML config for reference-free and reference-based workflows.
Shared parameters¶
General parameters¶
The first things we want to specify for QuiCAT to run are:
sequencing_technology: with this parameter we can specify which data are we dealing with:DNAif it is a bulk DNA dataset10xif it is a dataset from 10x genomics, like Chromium single cell or VisiumParseif it is a dataset from Parse biosciences
inputto specify whether we are givingfastqfiles orbamfiles as input.paired_endif it we are using fastq files input and it is from paired end sequencing.
QC parameters¶
Here we specify the criteria to retain the barcode based on filtering thresholds:
barcodes_path: By setting this to true, we can then provide QuiCAT with a set of whitelisted cells or spots. Reads coming from outside the whitelisted cells are automatically discarded.phred33: if set to true, it will use PHRED33 score for quality control otherwise it will use PHRED64read_qc_thresholdandread_qc_percentageused in combination allow QuiCAT to discard reads based on quality scores. In particular the user can set how many base pairs in percentage need to have a PHRED score of at least the specified threshold.filter_barcodes_relative_abundancediscard barcodes which frequencies are below the specified abundance threshold.filter_barcodes_raw_numbers: discard barcodes which absolute counts are below the specified threshold.
In our specific case we are dealing with a bulk DNA dataset with fastq input files and paired end sequencing. Furthermore we want to retain reads where at least 80% of the base pairs have a PHRED score of at least 20, and discard the barcodes that have a relative abundance below 0.001%.
The config file will therefore look like this:
config:
sequencing_technology: "DNA"
input: "fastq"
paired_end: true
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
Reference-Free parameters¶
Now that all the general parameters are set, it is time to set the Reference-Free specific parameters.
Using the flanking regions¶
First we start by specifying the reference itself. To tell QuiCAT that we want to use the Reference-Free approach, we can simply specify a string in the reference field. If we specify a path to a reference file in either FASTA, JSON, or TXT, then the reference-based approach is used instead.
Note that if we specify flanking regions, we can use:
- "*SEQUENCE" to specify the downstream region
- "SEQUENCE*" to specify the upstream region
- "SEQUENCE*SEQUENCE" to specify both
Given the structure of the modified LARRY barcode, we can use two strategies:
Either we specify the downstream adapter, or we specify the structure of the barcode indicating the conserved nucleotides and masking the others with 'N' We will start by creating a configuration file to use the downstream flanking region.
config:
...
reference: "*CATA"
Next, we need to define additional parameters that are specific to the Reference-Free approach.
If we are specifying flanking regions, then we need to add flanked_pattern: true; otherwise, we can omit this field.
We can then specify a range for the barcode length using min_read_length and max_read_length, or alternatively, specify a fixed barcode length with read_length.
The flanking_mismatches parameter allows the user to indicate whether they are willing to accept some mismatches in the flanking regions, or if they only want to extract barcodes that have a 100% match in those regions. If left blank or set to 0, QuiCAT will use REGEX matching to extract only barcodes with a perfect flanking match. If a value is instead specified, QuiCAT switches to CUTADAPT and can extract barcodes even when there are mismatches in the flanking regions.
For example, if we set flanking_mismatches: 0.25, we allow QuiCAT to extract barcodes even if up to 25% of the nucleotides in the specified flanking region are mismatched.
Finally, we want to specify whether to collapse barcodes with low counts into those with higher counts, in order to correct for sequencing errors. For this, we will use two parameters: distance_threshold and barcode_ratio. With distance_threshold, we tell QuiCAT to collapse barcodes with low counts into barcodes with higher counts, provided that their distance is below the specified threshold. With barcode_ratio, we instruct QuiCAT to perform the collapse only if the higher-count barcode is more frequent than the lower-count one by the specified factor.
For example, if we want to collapse low-frequency barcodes into high-frequency ones when their distance is within 2 base pairs of difference, and we want to retain low-frequency barcodes unless the higher-frequency ones are at least 3 times more abundant, then the config file will have:
In our case we want to use the downstream region CATA, allow 25% mismatch in the flanking region, and set the read length to 16 nucleotides.
Furthermore we want to collapse low frequency barcodes, so we set the distance threshold to 2, but we only want to collapse barcodes into high-frequency ones only if the high-frequency ones have frequencies of at least three times higher than the low frequency ones therefore setting the barcode ratio to 3.
To bring it all together, the final config file will look like this:
config:
sequencing_technology: "DNA"
input: "fastq"
paired_end: true
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
reference: "*CATA"
flanked_pattern: true
read_length: 16
flanking_mismatches: 0.25
distance_threshold: 2
barcode_ratio: 3
Using the barcode pattern¶
Some barcodes library like LARRY have conserved nucleotide positions that can also be leveraged for the extraction process.
Recalling the modified LARRY barcode structure ( NNNN CT NNNN AC NNNN ) we can use the conserved nucleotides and mask all the other positions with N
config:
...
reference: "NNNNCTNNNNACNNNN"
Since using this pattern we are already specifying the length of the barcodes, we do not need to use length parameters in this setup.
Likewise since the flanking region is not used, the flanking_mismatches parameter is also superfluous here.
The update version of the config file would therefore look like this:
config:
sequencing_technology: "DNA"
input: "fastq"
paired_end: true
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
reference: "NNNNCTNNNNACNNNN"
flanked_pattern: false
read_length: null
flanking_mismatches: null
distance_threshold: 2
barcode_ratio: 3
Reference-Based parameters¶
As mentioned above, to switch from the reference-free to the reference-based workflow, it is sufficient to specify the path to a reference file instead of a string in the reference parameter of the config file.
How to provide a reference to QuiCAT¶
QuiCAT accepts 3 file formats for the reference: FASTA, JSON, or plain TXT files. The key difference is that when using FASTA or JSON, the barcode name as reported in the reference file will be stored in the final output. If a plain TXT file is used, then the raw sequences themselves are stored in the final outputs.
Below we show an example with only two clone from this experiment on how to setup the different reference files.
FASTA example¶
To provide a reference in FASTA format, the user can create a FASTA file where each entry contains a barcode sequence to be extracted, and the header represents the name of the barcode as the user wants it to appear in the final output.
>clone_01
GTGCCTATGCACAAAA
>clone_02
TCGCCTAATGACTTAA
...
JSON example¶
To provide a reference in JSON format, the user can create a JSON file storing keys and values, where each key contains a barcode sequence to be extracted, and the corresponding values represents the name of the barcode as the user wants it to appear in the final output.
{
"GTGCCTATGCACAAAA": "clone_01",
"TCGCCTAATGACTTAA": "clone_02",
...
}
TXT example¶
To provide a reference in TXT format, the user can create a plain TXT file where each line represent a sequence, separated by the newline character.
GTGCCTATGCACAAAA
TCGCCTAATGACTTAA
...
Once again contrary to FASTA and JSON, if a TXT file is used, the raw sequences will be stored in the final output rather than the barcodes'names as specified by the user.
Specifying the remaining parameters¶
Most of the parameters used in the reference-free workflow are no longer required in the reference-based workflow. In particular, the parameters related to barcode length and barcode collapsing become superfluous.
Instead, the only relevant parameter is now aln_mismatches. As described in the manuscript—and similarly to the reference-free workflow—the reference-based workflow of QuiCAT leaves it to the user to decide whether to be error-tolerant or not.
If aln_mismatches is set to 0, the Aho-Corasick algorithm will be used to extract barcodes with a 100% match to the reference in almost linear time, at the expense of sensitivity. Conversely, if aln_mismatches is set to a value equal to or greater than 1, QuiCAT switches to the global aligner EDLIB.
With this setup, Aho-Corasick will be used:
config:
...
aln_mismatches: null or 0
Whereas to allow for mismatches in the alignment, the aln_mismatches parameter can be used to control how many mismatches are permitted:
config:
...
aln_mismatches: 1 or greater
The final config file will therefore look like this:
config:
sequencing_technology: "DNA"
input: "fastq"
paired_end: true
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
reference: "/path/to/reference/file"
aln_mismatches: ...
I/O¶
Finally the last two parameters to be set in the YAML file are the path to a csv file to specify sample metadata and relevant files path (see below), and the path to an output directory where to store the final outputs of the extraction workflow.
config:
...
input_csv: "/path/to/input.csv"
output_path: "/path/to/output/directory"
The input csv file¶
Next we have to create an input csv file that contains the information about the samples we want to process, and the path to the relevant files.
The input csv file should have the following columns:
sample,fastq_path_r1,fastq_path_r2,barcodes_path,bam_path,condition,replicate
In our case since we are working with fastq files, with paired end sequencing and no whitelisted cells are specified the columns that we should fill are sample,fastq_path_r1,fastq_path_r2, whereas barcodes_path,bam_path can be left blank.
The input csv file should look like this:
sample,fastq_path_r1,fastq_path_r2,barcodes_path,bam_path,condition,replicate
sample_01,/path/to/R1.fastq,/path/to/R2.fastq,,,,
condition and replicate are instead optional metadata fields that would be added in the final output metadata columns in case they are filled in.
Running the extraction workflow¶
Once both the YAML configuration file and the input csv file are created, running the extraction workflow is as easy as running on a terminal:
quicat extract /path/to/config.yaml
QuiCAT will first validate the config file, check if the files specified in the input csv file exist, and then start the extraction process.
Once it is done, QuiCAT will store the results at the specified location both in csv and anndata h5ad format which can be imported with scanpy for further downstream analyses.
Working with BAM files¶
So far we created a config file to work with fastq files but QuiCAT can also work with BAM files. Let's go back to the latest version of our config file we generated for our reference-based workflow:
config:
sequencing_technology: "DNA"
input: "fastq"
paired_end: true
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
reference: "/path/to/reference/file"
aln_mismatches: ...
To switch from FASTQ to BAM, all we have to do is change the input field from "fastq" to "bam". The paired_end parameter is now superfluous and can be set to null:
config:
input: "bam"
paired_end: null
...
Additionally, when working with BAM files, the user can choose whether to scan the entire file or focus on reads aligned to a particular contig by using the contig parameter. If the contig parameter is set to null, the whole file will be scanned:
config:
input: "bam"
paired_end: null
contig: null
...
If, for example, the user only wants to scan reads aligned to Chromosome 1, it can be specified as:
config:
input: "bam"
paired_end: null
contig: 'Chr1'
...
Lastly, if the user wants to focus on unaligned reads—which is often where genetic barcodes are found—this can be specified by setting contig to "*":
config:
input: "bam"
paired_end: null
contig: '*'
...
The rest of the parameters remain unchanged. If for example we still want to use the reference-based workflow as previously shown, but using BAM files as input instead while only scanning unmapped reads, the final config file will look like this:
config:
sequencing_technology: "DNA"
input: "bam"
paired_end: null
barcodes_path: false
phred33: true
read_qc_threshold: 20
read_qc_percentage: 80
filter_barcodes_relative_abundance: 0.001
filter_barcodes_raw_numbers: null
reference: "/path/to/reference/file"
aln_mismatches: ...
contig: "*"
input_csv: "/path/to/input.csv"
output_path: "/path/to/output/directory"
The csv file will need to be modified as well to specify the path to the BAM file while leaving the FASTQ fields empty:
sample,fastq_path_r1,fastq_path_r2,barcodes_path,bam_path,condition,replicate
sample_01,,,,/path/to/bam,,
FASTA file for reference-based run¶
Here is the fasta file with the complete reference to run this experiment using the reference-based approach
>clone_01
GTGCCTATGCACAAAA
>clone_02
TCGCCTAATGACTTAA
>clone_03
GGCCCTTAGCACAATC
>clone_04
CTAACTAGCGACGGAG
>clone_05
TTCACTGACTACGACT
>clone_06
CCTGCTGCTAACCTTT
>clone_07
TAACCTACGAACCAAG
>clone_08
CTTTCTCCTTACCCTG
>clone_09
AGCGCTTAGAACGCTT
>clone_10
TGTACTTACGACGGTA
>clone_11
CGGACTGGGGACGGCG
>clone_12
AGGTCTGTCTACATTG
>clone_13
AGGCCTGATCACGAAT
>clone_14
CGTACTTCCGACGTAC
>clone_15
CGTCCTTGTTACGTGG
>clone_16
TGTGCTTATGACTCGC
>clone_17
CATGCTGGGCACTGGG
>clone_18
CGTACTGGTGACGGGC
>clone_19
TCGTCTTAGTACTCCT
>clone_20
CAAACTATCCACGCCG
>clone_21
AACACTACTGACATTC
>clone_22
TCAACTTGGAACTTAG
>clone_23
TGAGCTAGCTACACAG
>clone_24
AAGACTATATACTAAT
>clone_25
GTGCCTGGTGACGATT
>clone_26
GTCCCTGATCACGTCG
>clone_27
GTTTCTAAATACGAGA
>clone_28
AAACCTCTATACGTTT
>clone_29
GATACTTTAGACGAAC
>clone_30
TGCACTAGTCACGGTT
>clone_31
AATGCTGTTAACGAGG
>clone_32
GTGTCTTATCACAGTG
>clone_33
TTTGCTATCAACTCGG
>clone_34
CTTACTGTACACGTAT
>clone_35
TTATCTGCATACACGT
>clone_36
TGGGCTCAGAACTGTC
>clone_37
TGAACTTGAGACATCG
>clone_38
CTCGCTGACGACCTCC
>clone_39
TACGCTCGTGACCCAG
>clone_40
AACTCTGTGAACTTAA
>clone_41
CACGCTGCTCACAGGC
>clone_42
TTAGCTGGGGACCCAT